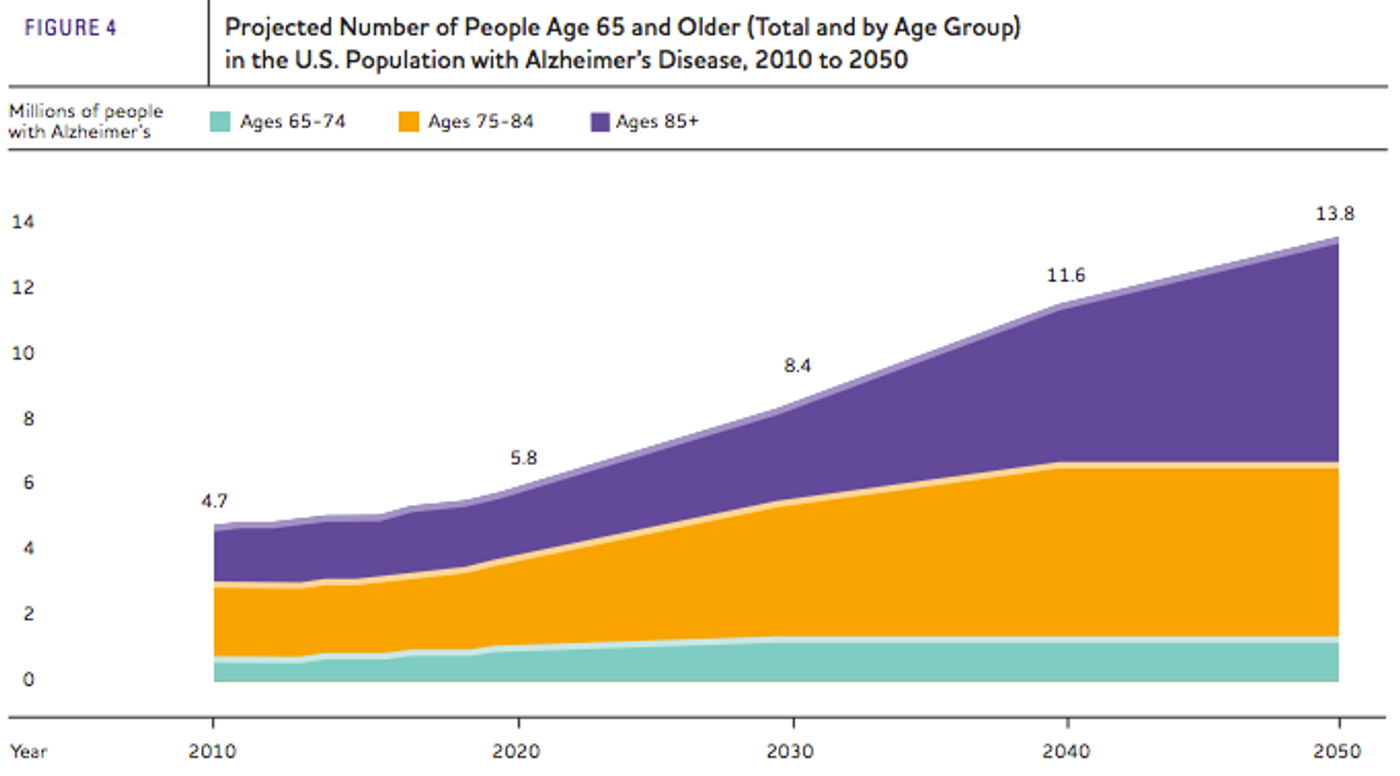
Alzheimer’s Disease (AD) is the most common form of dementia, currently affecting 5.5 million people in the U.S. There are currently no means of preventing or significantly slowing down disease progression, and unless there are some major advancements in the next few years, the number of people with AD in the U.S. will grow to 13.8 million by 2050.
Amyloid-β accumulation is the main pathological feature of AD and so most of the therapeutics designed and tested have been focused on the production or clearance of amyloid-β. Drugs that have been tested to reduce the production of amyloid-β include ?-secretase inhibitors and BACE1 inhibitors. BACE1 and ?-secretase are two of the enzymes that cleave the amyloid precursor protein into amyloid-β. Thus far, this strategy of targeting amyloid-β by targeting the proteases that create it has not been successful. With that said, most major pharmaceutical companies have a new generation type of BACE1 inhibitor somewhere in the pipeline, so there could be some exciting developments on the horizon. Anti-amyloid antibodies have been developed to enhance the clearance of amyloid-β from the brain. Again, there has been limited success with this strategy, but one antibody in particular, aducanumab, has had some success in the very early, prodromal stages of AD. Because there has been such limited success thus far, we are in need of novel therapeutic targets for the prevention of AD.
A recent paper published in
Science Translational Medicine describes a potential new method to enhance clearance of amyloid-β from the brain that could be targeted for AD therapeutics.
Heparan sulfate proteoglycans (HSPGs) are a type of very abundant cell membrane and extracellular space molecule that consist of a protein core covalently bonded to multiple heparan sulfate (HS) chains. HSPGs have been shown to bind to amyloid-β and accelerate its aggregation while HS alone has been implicated in cellular uptake of amyloid-β and in the mediation of amyloid-β neurotoxicity and inflammatory response. Because of these observations, Guojun Bu from the Mayo Clinic Department of Neuroscience led his team to investigate the interaction between HSPGs and amyloid-β metabolism.
Long story short, they discovered
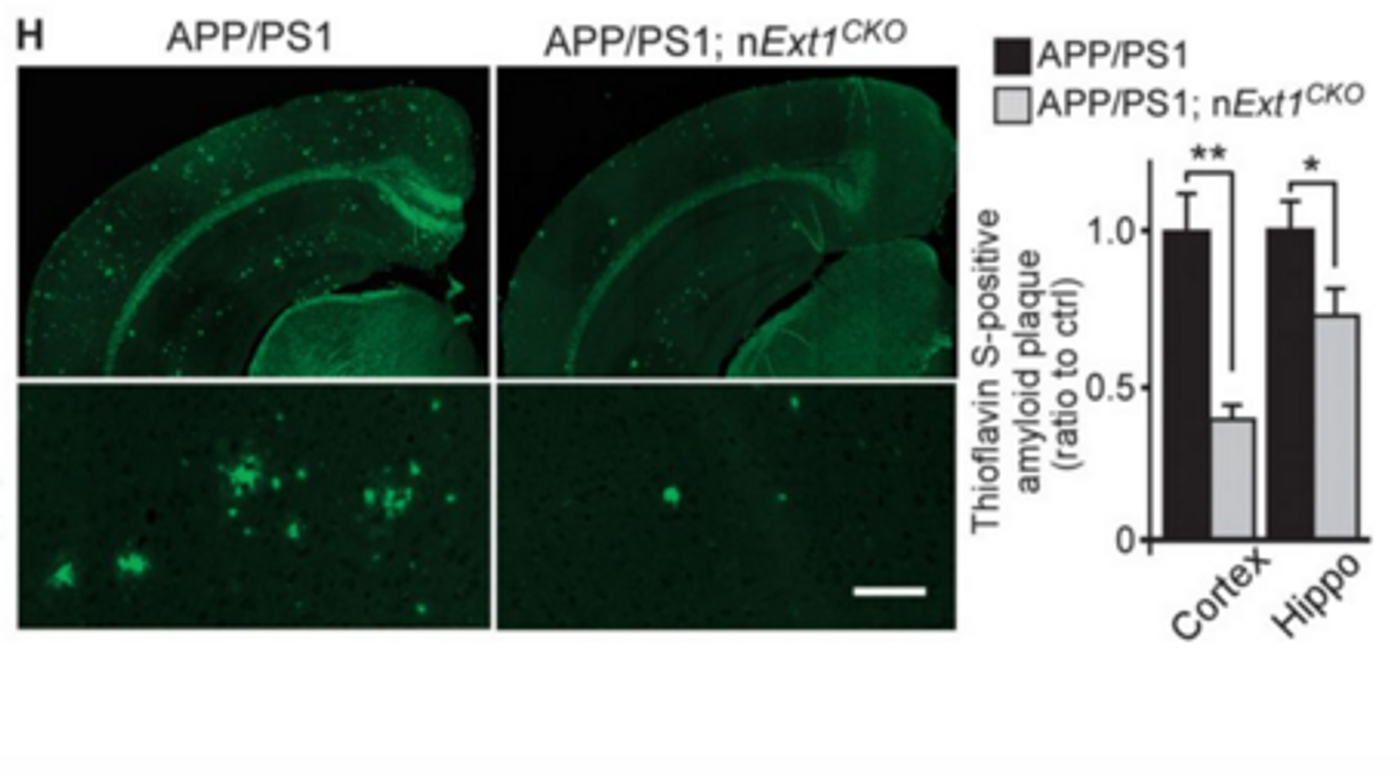
in vivo evidence that neuronal HS exacerbates amyloid plaque deposition and inhibits clearance of amyloid-β from the brain. To study the effects of HS, they created a conditional knockout mouse model using the APP/PS1 mouse model of AD. To get rid of HS and HSPG expression, the researchers knocked out the
Ext1 gene in adult forebrain neurons.
Ext1 encodes an enzyme that is required for the formation of HS, so without it, there are no HSPGs or HS chains. Using microdialysis to capture the real-time kinetics of amyloid-β in vivo, they saw that the mice lacking HS were able to clear amyloid-β from the brain faster than the APP/PS1 mice fully expressing HS. In addition, the conditional knockout APP/PS1 mice had significantly less amyloid deposition and neuroinflammation, as measured by activated microglia.
To confirm that these findings in mice is translatable to human AD pathology, the researchers also evaluated postmortem brain tissue from AD patients. They found that several HSPG species were upregulated compared to non-demented age-matched controls.
Amyloid pathology in AD is ultimately the result of an imbalance in amyloid-β processing and metabolism. The research presented here offers a new pathway to target for the treatment of AD. Inhibiting Ext1 could be a good target, or inhibiting the interaction between HS, HSPGs, and amyloid-β could also be exploited for drug development.