In a
study recently published in Science, researchers have described a cage they compare to the capsule structure that surrounds a virus, that they designed and created. These nanocages that were inspired by the molecular machines of nature, are capable of holding cargo like proteins or nucleic acids. An amazing thing about them is that they have compartments, and as such can assemble their contents in a controlled manner.
David Baker, a computational biochemist and lead author of the work, is revolutionizing science with his team. They have utilized new computational and genomic technologies to determine how amino acids fold into protein structures that are the building blocks of life, a major new achievement.
The scientists have gotten started by making new proteins meant to simultaneously fight every strain of influenza, an experimental vaccine for HIV, and even enzymes that enable microbes to absorb and convert atmospheric carbon dioxide.
Scientists have understood that proteins formed a 3D structure based on its amino acid sequence since the 1960s. However, it’s not known how to predict exactly what structure proteins will fold into based on their sequence alone.
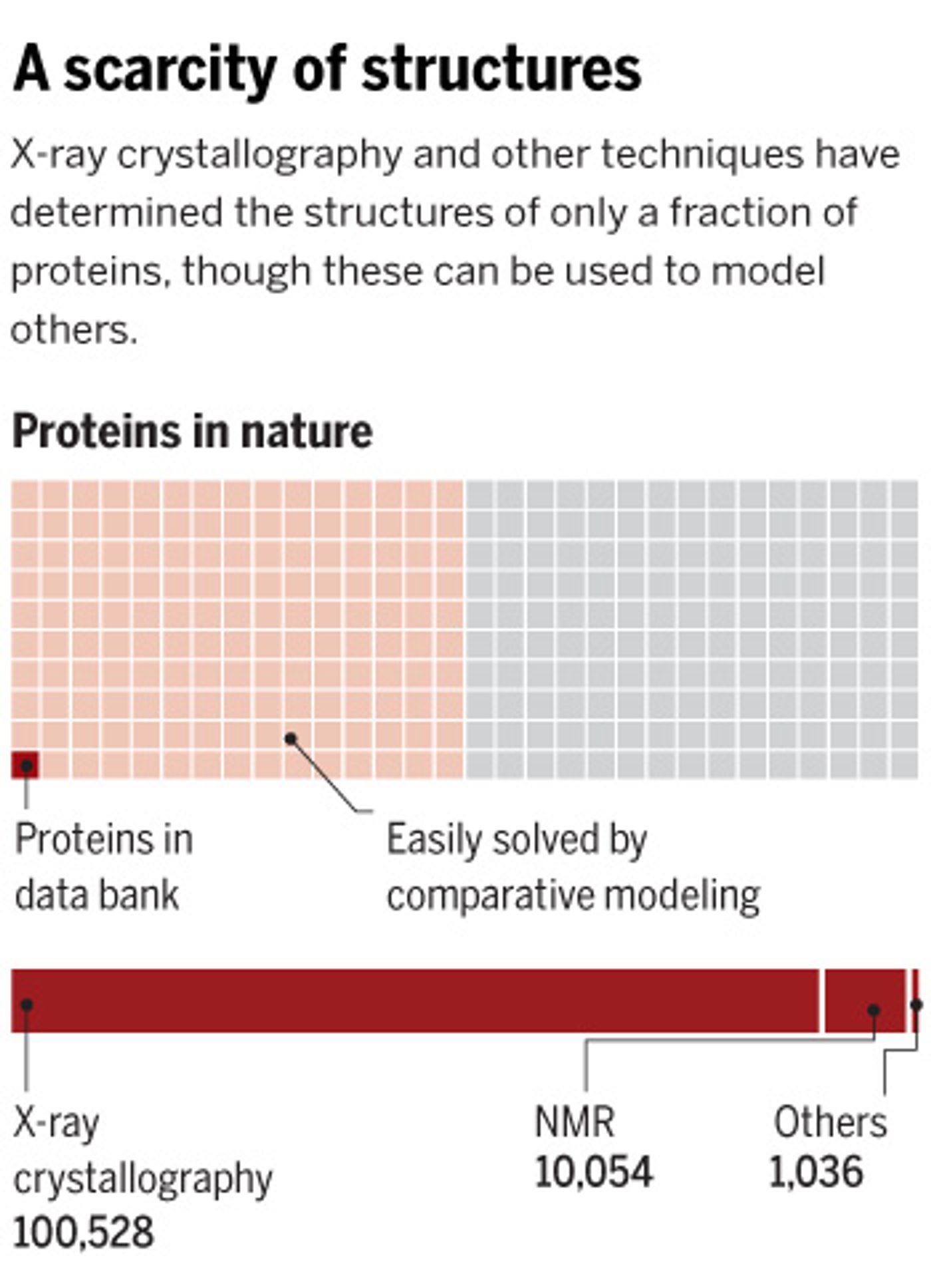
Protein structure can be determined using several techniques like nuclear magnetic resonance and x-ray crystallography. But of the millions of known proteins, only about 110,000 are contained in the international repository of such data, the
Protein Data Bank. That detail of knowledge about a protein can be critical to knowing its function, making it very important to biologists and chemists.
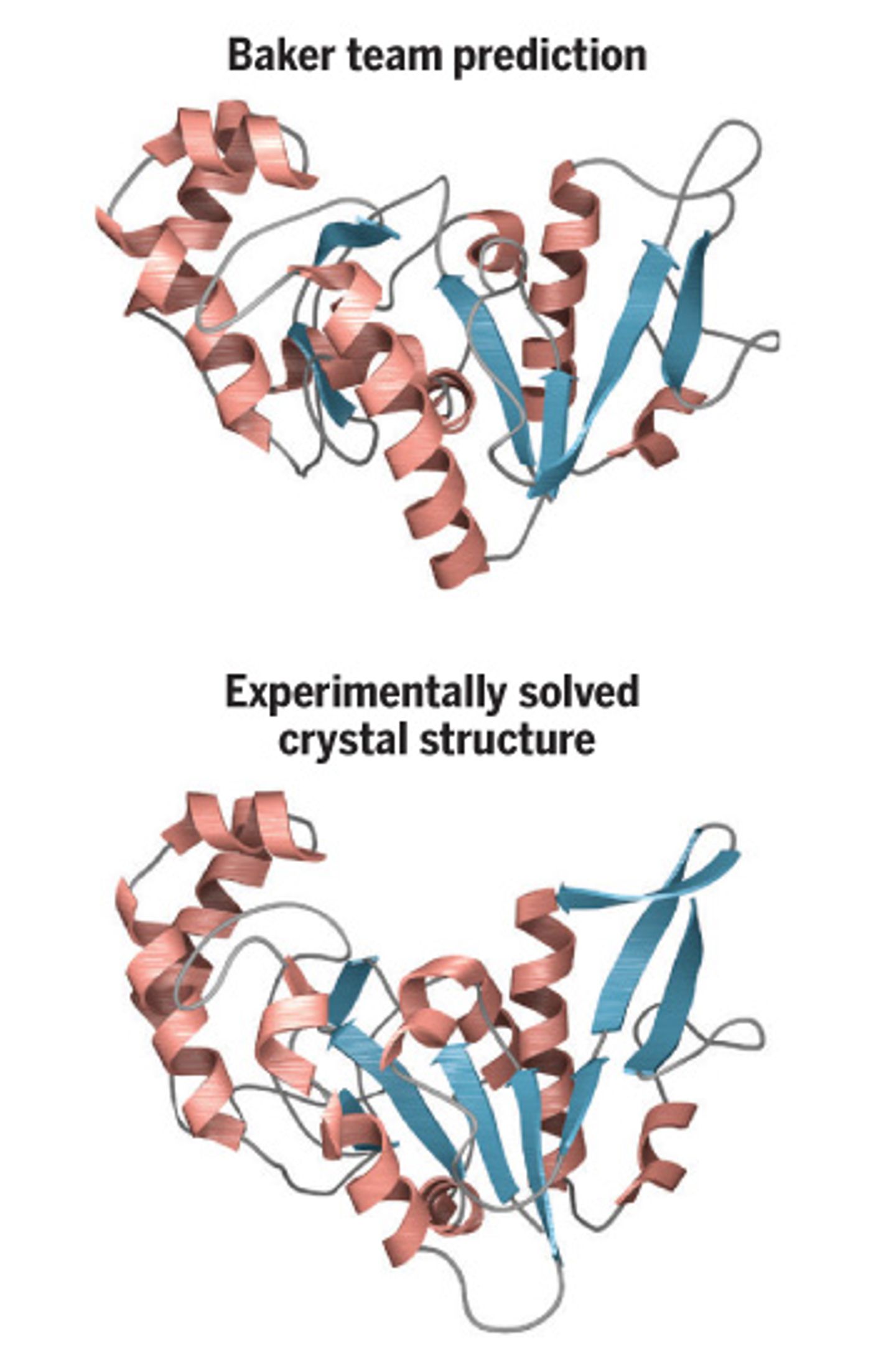
While models have been proposed, it took a program developed in Baker’s lab, Rosetta, to make the breakthrough. The work requires such immense computing power that the researchers have made a crowd funding extension -
Rosetta@home - which enables the public to give idle computer time to the venture. They’ve also created a video game where remote users can play around with protein folding -
fold it - and in turn, built a group of over a million members and about two dozen software packages.
“The most brilliant thing David has done is build a community,” Neil King, a former postdoctoral fellow in Baker’s lab, and currently an investigator at UW’s Institute for Protein Design (IPD), told Science.
Rosetta has limitations, and sometimes has struggled with accurately predicting the structure of larger proteins. “I wasn’t sure whether I would get there,” Baker said, “I don’t feel that way anymore.”
He was inspired by work done in the 1990s, when computational biologist Chris Sander, then with the European Molecular Biology Laboratory in Heidelberg, Germany, and now with Harvard, suggested that DNA sequences might help identify amino acids that would naturally pair together when proteins folded up.
Baker used the idea to create a new program that works in tandem with Rosetta, called Gremlin. The results have been nothing short of profound. Previous algorithms have worked out the structures of 56 proteins out of 8000 protein families for which there isn’t existing data. Since then, Baker’s and colleagues have added 900, and have big plans for the future.
Aside from looking at existing proteins, Baker wants to make proteins for a purpose. “We were limited by what existed in nature...we can now short-cut evolution and design proteins to solve modern-day problems,” he explains, “we can now build a whole new world of functional proteins.”
Sources:
Science